Phytochemicals and Molecular Docking: A Futuristic Approach for Drug Discovery
-
Atul Kaushik
School of Applied Chemistry & Basic Sciences, Sardar Bhagwan Singh University, Balawala, Dehradun, Uttarakhand, India
Jeevan Jyoti KaushikDepartment of Microbiology, School of Basic and Applied Sciences, Shri Guru Ram Rai University, Dehradun, Uttarakhand, India
Nand Lal

School of Life Sciences and Biotechnology, Chhatrapati Shahu Ji Maharaj University, Kanpur, Uttar Pradesh, India
| Received 16 May, 2025 |
Accepted 10 Aug, 2025 |
Published 30 Sep, 2025 |
The increasing prevalence of drug resistance and adverse effects associated with synthetic drugs has intensified the search for novel therapeutic agents from natural sources. Phytochemicals, bioactive compounds derived from plants, have gained renewed interest due to their diverse chemical structures and broad-spectrum pharmacological activities. Current researchers integrate traditional ethnobotanical knowledge with advanced computational techniques, particularly molecular docking, to expedite the drug discovery process. Molecular docking enables the prediction of interactions between phytochemicals and biological targets, streamlining the identification of potential drug candidates. Recent studies have successfully applied molecular docking to identify promising anti-cancer, anti-microbial, and anti-inflammatory agents from plants. Furthermore, docking combined with molecular dynamics simulations and in silico ADME (Absorption, Distribution, Metabolism, and Excretion) studies has improved the reliability of these findings. The application of artificial intelligence and machine learning in docking is also enhancing accuracy and predictive power. This review explores the synergy between phytochemicals and molecular docking, highlighting current research trends and emerging challenges. By bridging traditional plant-based therapeutics with modern computational tools, this approach offers a promising pathway for developing effective, safe, and affordable drugs to address global health challenges.
| Copyright © 2025 Kaushik et al. This is an open-access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
INTRODUCTION
The rise of antimicrobial resistance poses a significant threat to global health, as pathogens evolve and become increasingly resistant to existing treatments, leading to a high rate of treatment failures and mortality. The development of new antimicrobial agents that can combat drug-resistant organisms is urgently needed. The quest for novel therapeutic agents has entered a transformative era, largely driven by advancements in phytochemistry and computational biology. Phytochemicals, bioactive compounds derived from plants, have long been recognized for their potential medicinal properties, offering diverse mechanisms of action that could lead to effective treatments for various diseases1. As the complexity of drug interactions and target specificity becomes increasingly apparent, the integration of molecular docking techniques emerges as a pivotal strategy in drug discovery2. This computational approach allows researchers to predict the interaction between phytochemicals and biological targets at the molecular level, facilitating the identification of promising candidates for further development. The fusion of traditional phytochemical knowledge with modern technology is revolutionizing the field, enabling the discovery of innovative, nature-inspired solutions. A wide variety of protective phytochemicals in fruits, vegetables, whole grains, nuts, legumes, and herbal seasonings, the regular consumption of these foods is essential to ensuring a healthier population that has lower rates of heart disease and cancer3. The traditional drug development process, which typically spans 10-15 years, is both time-consuming and costly, often exceeding billions of dollars. Despite these investments, the failure rate remains high due to factors such as poor efficacy, adverse side effects, and the emergence of drug resistance, particularly in infectious diseases and cancers4. The growing burden of chronic diseases like cancer, diabetes, and neurodegenerative disorders demands innovative therapeutic approaches. Conventional synthetic drugs, while effective in many cases, can lead to adverse reactions, toxicity, and reduced efficacy over time. These challenges have intensified the need for discovering novel therapeutic agents with unique mechanisms of action and fewer side effects. In this context, natural products, especially phytochemicals, have emerged as a promising source of new drug candidates. Medicinal plants are incorporated in our day-to-day lives and help boost immunity and provide the best nutrition to fight against infections5. This study aims to evaluate the drug-likeness of phytochemicals by utilizing molecular docking approaches to identify their binding potential with target proteins, thereby supporting their role in futuristic drug discovery.
Importance of phytochemicals in modern medicine: Phytochemicals are naturally occurring bioactive compounds found in plants. These secondary metabolites, which include alkaloids, flavonoids, terpenoids, phenolics, and glycosides, serve various ecological functions such as defense against pathogens, UV protection, and pollinator attraction6. Over centuries, traditional systems of medicine like Ayurveda, Traditional Chinese Medicine (TCM), and African traditional medicine have utilized plants to treat a wide range of diseases. The scientific validation of these traditional practices has led to the discovery of many lead compounds for modern drug development. Phytochemicals are valued for their structural diversity and biological activity, which provide a vast chemical library for drug discovery7. Unlike synthetic compounds, phytochemicals are often more biocompatible and may exhibit multi-target mechanisms of action. This polypharmacology is particularly useful for treating complex diseases like cancer, where targeting multiple pathways simultaneously can improve therapeutic outcomes8.
Natural compound classes and their translational potential in drug development: Medicinal plants are well-established sources of diverse bioactive phytochemicals, including flavonoids, alkaloids, terpenoids, glycosides, and phenolic compounds. These natural constituents have long been utilized in traditional medicine and are increasingly investigated for their therapeutic potential using modern scientific tools. Among these, molecular docking has emerged as a powerful in silico technique for identifying lead compounds by simulating their binding affinities with specific biomolecular targets. Recent studies have demonstrated the antimicrobial potential of Alseodaphne andersonii, supported by significant inhibitory activity against multiple pathogenic bacterial strains9. Similarly, the roots of Tectona grandis have shown promising antitussive effects in in vivo models, validating their traditional use10. The antioxidant capacity and CNS-stimulant properties of Diplazium esculentum have also been confirmed through FRAP assays and behavioral studies11. Additionally, Cyphostemma adenocoule has exhibited substantial phospholipase A2 inhibitory activity, reinforcing its Ethnomedicinal application in the management of snake envenomation12. Furthermore, recent molecular docking-based screening identified a novel inhibitor of Cytosolic Phospholipase A2 (cPLA2) with a docking score superior to the known inhibitor ATK. This compound also demonstrated favorable ADME, drug-likeness, and toxicity profiles, making it a promising candidate for further structure-activity relationship (SAR) optimization and preclinical evaluation as an anti-epileptic agent13. These findings collectively highlight the value of integrating traditional botanical knowledge with computational tools to accelerate the discovery and development of phytochemical-based therapeutics targeting infectious, inflammatory, and neurological disorders.
|
Role of computational tools in drug discovery: In the past two decades, computational tools have become integral to the drug discovery pipeline. Molecular docking, molecular dynamics simulations, and in silico ADME/Toxicity predictions help streamline the identification of potential drug candidates by reducing the need for extensive in vitro and in vivo testing. These tools allow researchers to predict the binding affinity of compounds to target proteins, understand molecular interactions at an atomic level, and optimize lead compounds before synthesis and testing14. Molecular docking, in particular, has emerged as a powerful tool for identifying potential therapeutic agents from large libraries of phytochemicals. Docking can predict the stability, binding mode, and potential efficacy of the compound. This approach accelerates the identification of promising leads, reduces costs, and allows for the exploration of compounds that may be challenging to synthesize in a laboratory setting15.
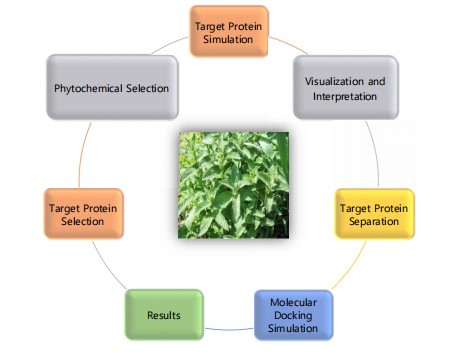
Molecular docking and dynamics study of phytochemicals: Given the convergence of traditional knowledge, phytochemical diversity, and computational advancements, the integration of phytochemicals and molecular docking (Fig. 1) offers a futuristic and promising approach to drug discovery. Numerous plant-derived phenolic compounds have exhibited wide-ranging biological activities of medicinal importance. Some notable examples include the stilbenoid resveratrol, Curcumin, Quercetin etc, which has shown numerous potential health benefits, including anti-carcinogenic activity (Table 1).
The process of phytochemical molecular docking involves several key steps aimed at understanding how natural compounds interact with biological targets. First, phytochemical selection involves identifying bioactive compounds from natural sources. Once selected, the preparation of the phytochemical follows, where the compound's structure is converted into a 3D format and optimized using molecular modeling software. The next step is target protein selection, where a relevant biological protein (such as a receptor or enzyme) is chosen, and its 3D structure is obtained, often from resources like the Protein Data Bank.
| Table 1: | Phytochemicals and their molecular docking studies | |||
| Phytochemical | Source plant | Target protein | Binding energy (kcal/mol) |
Potential application |
References |
| Curcumin | Curcuma longa | NF-κB (p65 subunit) | -5.42 | Anti-inflammatory, Anti-cancer |
Cheemanapalli et al.16 |
| Quercetin | Allium cepa, Citrus spp. |
SARS-CoV-2 3CL protease |
-8.58 | Antiviral (COVID-19) | Gasmi et al.17 |
| Berberine | Berberis vulgaris | HSD11B1 | -8.7 | Antipsychotic-induced metabolic syndrome |
Huang and Liu18 |
| Resveratrol | Vitis vinifera | MeCP2 | -94.76 | Cancer inhibition pathway |
Sahu et al.19 |
| Epigallocatechin Gallate (EGCG) |
Camellia sinensis | NLRP3 | -9.6 | Cellular injury | Jena et al.20 |
| Taxol (Paclitaxel) | Taxus brevifolia | β-Tubulin | -10.2 | Anti-cancer (Mitotic inhibitor) |
Yang et al.21 |
| Betulinic acid | Betula alba | Autocrine Motility Factor Receptor |
-7.22 | Refractory tumors | Saeed et al.22 |
| Ginsenoside- Rb1 & Rb2 |
Panax ginseng | BACE1 | -10 | Alzheimer’s disease | Choi et al.23 |
| Hesperidin | Citrus sinensis | PLK1 and EGFR | -9 | Cytoprotective | Rizvi et al.24 |
| Allicin | Allium sativum | FtsZ | -10 | Antibacterial, Antimicrobial |
Cahayani et al.25 |
| Piperine | Piper nigrum | TRPV1 (Transient receptor potential channel) |
-7.9 | Analgesic, Anti-inflammatory |
Karunakar et al.26 |
| Table 2: | Future perspectives on phytochemicals and molecular docking in drug discovery | |||
| Future perspective | Description | Potential impact | Major software/platforms |
| AI and machine learning integration |
AI to predict binding affinities and pharmacokinetics of phytochemicals |
Accelerates screening |
DeepDock, AlphaFold, DeepChem, AutoQSAR |
| Hybrid drugs (phytochemical and synthetic) |
Combining phytochemicals with synthetic drugs to improve efficacy and reduce side effects |
Enhances therapeutic potential through synergistic action |
AutoDock, AutoDock Vina, Molecular Operating Environment (MOE) |
| Personalized medicine | Tailoring phytochemical therapies based on individual genetic profiles |
Optimizes treatment outcomes for specific populations |
OpenEye, Schrödinger’s Maestro, DockThor |
| Nanotechnology-based delivery |
Developing nanoparticles and liposomes to enhance phytochemical bioavailability |
Improves stability, targeted delivery, and therapeutic efficacy |
NanoDDS, Arguslab, GROMACS, LAMPPS |
| Focus on neglected diseases | Identifying phytochemicals for treating rare and neglected diseases |
Addresses gaps in research for underserved medical conditions |
PyRx, iGEMDOCK, Docking Server |
| Multi-target drug discovery | Identifying phytochemicals that interact with multiple molecular targets |
Effective for treating complex diseases like cancer and neurodegeneration |
AutoDock Vina, Glide (Schrödinger), FlexX |
| In-Silico toxicity and ADMET prediction |
Using computational tools to predict safety and pharmacokinetics of phytochemicals |
Enhances safety profiles and reduces preclinical failures |
Swiss ADME, pkCSM, ADMET Predictor |
| Virtual reality (VR) in docking | Using VR for 3D visualization of phytochemical-protein interactions |
Improves understanding and design of targeted therapies |
Nanome, Covalent docking with ChimeraX, VR-Vina |
| Expanding Plant Databases | Growing phytochemical databases with new plant species and compounds |
Facilitates the discovery of novel therapeutic agents |
ChEMBL, PubChem, IMPPAT, NPASS |
In protein preparation, the protein structure is cleaned by removing water molecules and ligands, and hydrogen atoms are added to correct the charges. The docking setup phase defines the binding site on the target protein, and docking parameters are chosen, followed by the use of suitable software for the simulation. The molecular docking simulation predicts how the phytochemical interacts with the protein by running the docking algorithm. Afterward, results analysis is carried out to evaluate the binding energy scores and to examine hydrogen bonding and other molecular interactions. The results are then visualized and validated using software like PyMOL or Chimera, ensuring the docking predictions align with experimental data. Finally, the findings are reported and interpreted, with recommendations for possible therapeutic applications of the phytochemical based on its binding potential.
FUTURE PERSPECTIVES
Future outlooks point to a revolutionary change in pharmacological methods as scientists continue to investigate the enormous potential of phytochemicals and molecular docking in drug discovery. The identification and optimization of lead compounds from natural sources could be made easier by combining cutting-edge computational methods with phytochemical studies. Scientists may make previously unheard-of predictions about how bioactive phytochemicals and target proteins will interact by using molecular docking simulations. In addition to increasing the effectiveness of medication design procedures, this collaboration opens the door for the development of new therapeutic medicines with fewer adverse effects. Furthermore, the growing emphasis on personalized medicine reinforces the need for tailored phytochemical applications, which can be achieved through targeted molecular docking studies. Collectively, these advancements signal a promising future where the amalgamation of phytochemical research and cutting-edge technology revolutionizes drug discovery paradigms, ultimately benefiting global health (Table 2).
CONCLUSION
In conclusion, the integration of phytochemicals with molecular docking represents a transformative approach in drug discovery, particularly in identifying novel therapeutic agents against complex diseases such as cancer. The findings underscore the therapeutic potential of various phytochemicals, such as Kushenol T and Neocalyxins A, which exhibit strong binding affinities to critical proteins like CDK2, a key player in cancer progression. Furthermore, the utility of advanced computational methods not only accelerates the identification of these bioactive compounds but also enhances their specificity and efficacy, as illustrated by the exploration of phytochemicals targeting the Bile Salt Export Pump (BSEP) in cholestasis treatment.This dual focus on phytochemicals and molecular docking paves the way for more effective, safe, and individualized therapies, heralding a new era in pharmacology that aligns with the urgent need for innovative solutions in healthcare.
SIGNIFICANCE STATEMENT
The application of advanced computational methods not only expedites the identification process but also enhances the efficacy and specificity of various bioactive compounds. This simultaneous focus on phytochemicals and molecular docking opens the way to safer, more customized, and more effective medicines by predicting which phytochemicals are likely to demonstrate efficacy, especially in novel or developing disorders. Additionally, it marks the beginning of a new age in pharmacology that aligns with the urgent need for innovative solutions in the medical field.
ACKNOWLEDGMENT
The authors are thankful to the people who are directly or indirectly involved in the preparation of the manuscript.
REFERENCES
- Nahar, L. and S.D. Sarker, 2024. Computational Phytochemistry: An Overview. In: Computational Phytochemistry, Sarker, S.D. and L. Nahar (Eds.), Elsevier, Amsterdam, Netherlands, ISBN: 978-0-443-16102-5, pp: 1-58.
- Lin, X., X. Li and X. Lin, 2020. A review on applications of computational methods in drug screening and design. Molecules, 25.
- Craig, W.J., 1997. Phytochemicals: Guardians of our health. J. Am. Diet. Assoc., 97: S199-S204.
- Petrova, E., 2013. Innovation in the Pharmaceutical Industry: The Process of Drug Discovery and Development. In: Innovation and Marketing in the Pharmaceutical Industry: Emerging Practices, Research, and Policies, Ding, M., J. Eliashberg and S. Stremersch (Eds.), Springer, New York, ISBN: 978-1-4614-7801-0, pp: 19-81.
- Singh, G., P. Sahu, A. Sharma, B. Butool, R. Sarkar, R. Mishra and R. Soni, 2022. Public awareness about the usage of medicinal herbs found in the kitchen and their potential against COVID-19 disease. Plant Arch., 22: 288-296.
- Bidlack, W.R. and W. Wang, 2000. Designing Functional Foods to Enhance Health. In: Phytochemicals as Bioactive Agents, Bidlack, W.R., S.T. Omaye, M.S. Meskin and D.K.W. Topham (Eds.), CRC Press, Boca Raton, Florida, ISBN: 9780429181252, pp: 241-270.
- Ansari, S., 2021. Overview of Traditional Systems of Medicine in Different Continents. In: Preparation of Phytopharmaceuticals for the Management of Disorders: The Development of Nutraceuticals and Traditional Medicine, Egbuna, C., A.P. Mishra and M.R. Goyal, Elsevier, Amsterdam, Netherlands, ISBN: 978-0-12-820284-5, pp: 431-473.
- Macht, D.I., 1930. Contributions to phytopharmacology or the applications of plant physiology to medical problems. Science, 71: 302-306.
- Parcha, V., A. Kaushik, J.J. Kaushik, M.S.M. Rawat, S.A.S. Biswas, 2007. Evaluation of antimicrobial potential of Alseodaphne andersonii. leaf extracts against pathogenic bacteria. Pharm. Biol., 45: 60-63.
- Kaushik, A., M. Kumari, and A. Ambesajir, 2011. Studies on antitussive effect of Tectona grandis roots using a cough model induced by sulfur dioxide gas in guinea pigs. Int. J. Phytomed., 3: 279-284.
- Kaushik, A., C. Jijta, J.J. Kaushik, R. Zeray, A. Ambesajir and L. Beyene, 2012. FRAP (ferric reducing ability of plasma) assay and effect of Diplazium esculentum (Retz) Sw. (a green vegetable of North India) on central nervous system. Indian J. Nat. Prod. Resour., 3: 228-231.
- Kaushik, A., T.S. Tesfai, D.K. Barkh, F.K. Ghebremeskel, H.G. Zerihun and S.W. Woldeab, 2020. Evaluation of snake venom’s phospholipaseA2 enzyme inhibition activity of Cyphostemma adenocoule. Curr. Chem. Biol., 14: 196-202.
- Kaushik, A., H.A. Abu, I.Y. Tsegay, J.G.H. G/Michael, L.S. Habtesion and Y.F. G/Michael, 2025. Novel anti-epileptic drug design targeting the brain capillary leakage pathway; an in silico approach. Sci. Digest, 1: 16-17.
- Leelananda, S.P. and S. Lindert, 2016. Computational methods in drug discovery. Beilstein J. Org. Chem., 12: 2694-2718.
- Basu, A., A. Sarkar and U. Maulik, 2020. Molecular docking study of potential phytochemicals and their effects on the complex of SARS-CoV2 spike protein and human ACE2. Sci. Rep., 10.
- Cheemanapalli, S., N. Chinthakunta, N.M. Shaikh, V. Shivaranjani, R.R. Pamuru and S.K. Chitta, 2019. Comparative binding studies of curcumin and tangeretin on up-stream elements of NF-kB cascade: A combined molecular docking approach. Network Model. Anal. Health Inf. Bioinf., 8.
- Gasmi, A., P.K. Mujawdiya, R. Lysiuk, M. Shanaida and M. Peana et al., 2022. Quercetin in the prevention and treatment of coronavirus infections: A focus on SARS-CoV-2. Pharmaceuticals, 15.
- Huang, Z. and X. Liu, 2022. Network pharmacology and molecular docking analysis on targets and mechanisms of berberine in atypical antipsychotic-induced metabolic syndrome. Nat. Prod. Commun., 17.
- Sahu, R.K., V.V. Verma, A. Kumar, S. Tandon, B.C. Das and S.T. Hedau, 2022. In silico prediction and interaction of resveratrol on methyl-CpG binding proteins by molecular docking and MD simulations study. RSC Adv., 12: 11493-11504.
- Jena, A.B., U.C. Dash and A.K. Duttaroy, 2022. An in silico investigation on the interactions of curcumin and epigallocatechin-3-gallate with NLRP3 Inflammasome complex. Biomed. Pharmacother., 156.
- Yang, Y., A.A. Alcaraz and J.P. Snyder, 2009. The tubulin-bound conformation of paclitaxel: T-Taxol vs “PTX-NY”. J. Nat. Prod., 72: 422-429.
- Saeed, M.E.M., N. Mahmoud, Y. Sugimoto, T. Efferth and H. Abdel-Aziz, 2018. Betulinic acid exerts cytotoxic activity against multidrug-resistant tumor cells via targeting autocrine motility factor receptor (AMFR). Front. Pharmacol., 9.
- Choi, R.J., A. Roy, H.J. Jung, M.Y. Ali and B.S. Min et al., 2016. BACE1 molecular docking and anti-Alzheimer's disease activities of ginsenosides. J. Ethnopharmacol., 190: 219-230.
- Rizvi, S.M.D., M.P. Mudagal, S.S. Boregowda, T. Hussain and T. Al Hagbani et al., 2023. The flavonoid hesperidin methyl chalcone as a potential therapeutic agent for cancer therapy: Molecular docking, in vitro cytotoxicity, and in vivo antitumor activity. Arabian J. Chem., 16.
- Cahayani, W.A., C. Tanuwijaya, L.X. Chi and Y. Mulyastuti, 2019. Antibacterial activity of garlic (Allium sativum) extract and molecular docking studies of allicin. AIP Conf. Proc., 2108.
- Karunakar, P., V. Krishnamurthy, C.R. Girija, V. Krishna, D.E. Vasundhara, N.S. Begum and A.A. Syed, 2012. In silico docking analysis of piperine with cyclooxygenases. J. Biochem. Tech., 3: S122-S127.
How to Cite this paper?
APA-7 Style
Kaushik,
A., Kaushik,
J.J., Lal,
N. (2025). Phytochemicals and Molecular Docking: A Futuristic Approach for Drug Discovery. Trends in Pharmacology and Toxicology, 1(1), 49-55. https://doi.org/10.21124/tpt.2025.49.55
ACS Style
Kaushik,
A.; Kaushik,
J.J.; Lal,
N. Phytochemicals and Molecular Docking: A Futuristic Approach for Drug Discovery. Trends Pharm. Toxicol. 2025, 1, 49-55. https://doi.org/10.21124/tpt.2025.49.55
AMA Style
Kaushik
A, Kaushik
JJ, Lal
N. Phytochemicals and Molecular Docking: A Futuristic Approach for Drug Discovery. Trends in Pharmacology and Toxicology. 2025; 1(1): 49-55. https://doi.org/10.21124/tpt.2025.49.55
Chicago/Turabian Style
Kaushik, Atul, Jeevan Jyoti Kaushik, and Nand Lal.
2025. "Phytochemicals and Molecular Docking: A Futuristic Approach for Drug Discovery" Trends in Pharmacology and Toxicology 1, no. 1: 49-55. https://doi.org/10.21124/tpt.2025.49.55

This work is licensed under a Creative Commons Attribution 4.0 International License.